Porucha primární hemostázy
Sem patří poruchy cévní stěny (Ehler-Danlosův syndrom s poruchou kolagenu), trombocytopenie (HIT) nebo trombocytopatie (Von Willebrandova choroba sekundárně, Bernard-Soulierův syndrom).

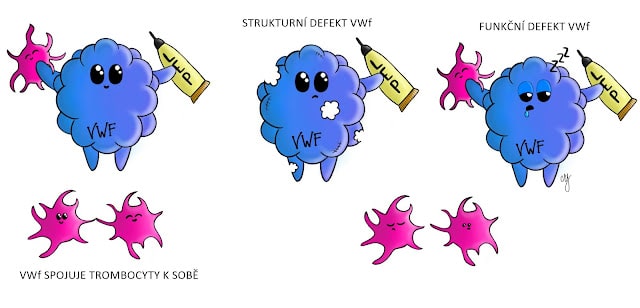
Von Willebrandova choroba (VWch)
Von Willebrandova choroba je způsobena defektem Von Willebrandova faktoru (VWf), který je potřebný pro adhezi a agregaci destiček. Defekt může být strukturní (v struktuře toho proteinu) nebo funkční. Nemoc je autosomálně dědičná, dle typu dominantně nebo recesivně.

Von Willebrandův faktor je tvořený sérii multimerů (2-100 jednotek VWf), které jsou uskladněny v endoteliálních buňkách a v megakaryocytech v α-granulích. Z trombocytů se uvolňuje pouze při jejich aktivaci.

Pro správnou adhezivní funkci jsou nezbytné multimery. Jejich balancovanou hladinu udržuje metaloproteáza ADAMTS13, jež štěpí nadměrně velké formy von Willebrandova faktoru na menší multimery. Těžký nedostatek ADAMTS13 (vrozený nebo získaný) je příčinou trombotické trombocytopenické purpury (TTP). Hemolyticko-uremický syndrom (HUS) má jinou etiopatogenezi (např. Shiga toxin, poruchy komplementu), přestože se u některých případů jejich mechanizmy mohou překrývat
Klinicky je patrné spíše slizniční krvácení a u žen s těžkým defektem může dojít k potratům v prvním trimestru.
Klasifikace

Diagnostika se zaměřuje na vyšetření VWf a krvácivé projevy v anamnéze. Screeningově můžeme vyšetřit počet trombocytů a APTT (prodloužený).
Léčba
Při léčbě se využívá podávání plazmatických koncentrátu FVIII a VWf. Antifibrinolytika (tranexamová kyselina) jsou doporučena k léčbě slizničního krvácení ve stomatologii.
Hemofilie
Hemofilie je vrozená krvácivá choroba, při které je v plazmě snížený faktor VIII (hemofilie A) nebo faktor IX (hemofilie B). Geny pro přenos hemofilie jsou lokalizovány na pohlavním chromozomu X.

Vzhledem k tomu, že hemofilie je gonosomálně recesivní, jsou ženy spíše přenašečky, než že by nemocí samy trpěly.
Klinicky můžeme hemofilii rozdělit dle aktivity faktoru na:

Typicky se hemofilie projevuje opožděným krvácením po zákrocích, snadnou tvorbou hematomů, krvácením po extrakci zubů, krvácení do zažívacího traktu, iliopsoatu či mozku. V důsledku kloubního krvácení dochází k synovitidě a destrukci kloubní chrupavky. U ženy přenašečky hrozí krvácení při poklesu faktoru pod 25%.

Diagnosticky je prodloužené APTT (Aktivovaný parciální tromboplastinový čas) poklesu fVIII nebo fIX pod 30 %. U každého hemofilika je také nutné vyloučit inhibitor (protilátky proti koagulačnímu faktoru) a alespoň 2x vyšetřit koagulační aktivitu fVIII a fIX. Po stanovení diagnózy je vhodné genetické vyšetření pro identifikaci přenašeček v rodině.
Léčbou hemofilie je substituce krevními deriváty. U hemofilie A podáváme fVIII, kdy 1 jednotka zvýší aktivitu faktoru VIII o 2%. Při hemofilii B podáváme fIX, kdy 1 jednotka zvedne aktivitu o 1%. Jako podpůrnou léčbu lze zvolit antifibrinolytika.
V případě vzniku inhibitoru, jako reakce imunitního systému na cizorodou látku, se podle imunitní odpovědi rozhodneme, zda zvýšíme dávky požadovaného faktoru (k vyvázání inhibitoru) nebo přejdeme na rekombinantní aktivovaný FVIIa, popřípadě koncentrát protrombinového komplexu.
Je také možnost pokusit se inhibitor eradikovat pomocí imunosupresivní léčby.
Inhibitor může vznikat i proti ostatním koagulačním faktorům. Přestože je tento stav vzácný, mělo by se diferenciálně diagnosticky takovýto specifický inhibitor vyloučit. Následkem léčby mohou být i trombózy v případě předávkování.
MUDr. Jan Málek
Máš otázky? Kontaktuj Honzu: TADY
Chci si udělat kvíz – TADY



